

Allele Specific Pcr Primer Design Tool
Primer Design Algorithms, Databases & Resources
- Real-time PCR primer & probe databases
- RTPrimerDB: the Real-Time PCR primer and probe database
- RTPrimerDB: the real-time PCR primer and probe database, major update 2006
- RTPrimerDB: the portal for real-time PCR primers and probes
- The BiSearch web server
- BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes
- PRaTo: A web-tool to select optimal primer pairs for qPCR
- AutoDimer: a screening tool for primer-dimer and hairpin structures
- A PCR primer bank for quantitative gene expression analysis
- A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance
- PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification
- PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update
- GETPrime: a gene- or transcript-specific primer database for quantitative real-time PCR
- methBLAST and methPrimerDB: web-tools for PCR based methylation analysis
- AtRTPrimer: database for Arabidopsis genome-wide homogeneous and specific RT-PCR primer-pairs
- Real-time PCR primer design tools
- RExPrimer: an integrated primer designing tool increases PCR effectiveness by avoiding 3' SNP-in-primer and mis-priming from structural variation
- BatchPrimer3: a high throughput web application for PCR and sequencing primer design
- Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analysis
- ConservedPrimers 2.0: a high-throughput pipeline for comparative genome referenced intron-flanking PCR primer design and its application in wheat SNP discovery
- PriSM: a primer selection and matching tool for amplification and sequencing of viral genomes
- PCRTiler: automated design of tiled and specific PCR primer pairs
- maxAlike: maximum likelihood-based sequence reconstruction with application to improved primer design for unknown sequences
- PrimerIdent: A web based tool for conserved primer design
- ThermoPhyl: a software tool for selecting phylogenetically optimized conventional and quantitative-PCR taxon-targeted assays for use with complex samples
- WASP: a Web-based Allele-Specific PCR assay designing tool for detecting SNPs and mutations
- Advanced qPCR design software
- Standarised primer optimisation and design specification. Application Note by National Genetics Reference Laboratory - Wessex
- BioInformatics in qPCR
- BioStatisctics in real-time PCR
- Determination of real-time PCR efficiency - NEW PAPERS
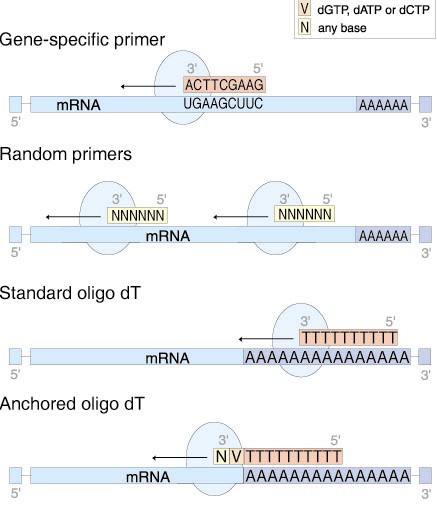
General Priming Strategies
Real-time PCR primer and probe databases:
- RTPrimerDB => http://medgen.ugent.be/rtprimerdb/ http://www.rtprimerdb.org

- RTPrimerDB: the Real-Time PCR primer and probe database
PATTYN, F., SPELEMAN, F., DE PAEPE A. & VANDESOMPELE, J.
Nucleic Acids Research, 31(1): 122-123The real-time polymerase chain reaction (PCR) methodology has become increasingly popular for nucleic acids detection and/or quantification. As primer/probe design and experimental evaluation is time-consuming, we developed a public database application for the storage and retrieval of validated real-time PCR primer and probe sequence records. The integrity and accuracy of the data are maintained by linking to and querying other reference databases. RTPrimerDB provides free public access through the Web to perform queries and submit user based information. Primer/probe records can be searched for by official gene symbol, nucleotide sequence, type of application, detection chemistry, LocusLink or Single Nucleotide Polymorphism (SNP) identifier, and submitter's name. Each record is directly linked to LocusLink, dbSNP and/or PubMed to retrieve additional information on the gene/SNP for which the primers/probes are designed. Currently, the database contains primer/probe records for human, mouse, rat, fruit fly and zebrafish, and all current detection chemistries such as intercalating dyes (SYBR Green I), hydrolysis probes (Taqman), adjacent hybridizations probes and molecular beacons. Real-time PCR primer/probe records are available at http://www.realtimeprimerdatabase.ht.st
- RTPrimerDB: the real-time PCR primer and probe database, major update 2006
Pattyn F, Robbrecht P, De Paepe A, Speleman F, Vandesompele J.
Center for Medical Genetics Ghent (CMGG), Ghent University Hospital, De Pintelaan 185, 9000 Ghent, Belgium.
Nucleic Acids Res. 2006 Jan 1;34(Database issue): D684-688The RTPrimerDB (http://medgen.ugent.be/rtprimerdb) project provides a freely accessible data retrieval system and an in silico assay evaluation pipeline for real-time quantitative PCR assays. Over the last year the number of user submitted assays has grown to 3500. Data conveyance from Entrez Gene by establishing an assay-to-gene relationship enables the addition of new primer assays for one of the 1.5 million different genes from 2300 species stored in the system. Easy access to the primer and probe data is possible by using multiple search criteria. Assay reports contain gene information, assay details (such as oligonucleotide sequences, detection chemistry and reaction conditions), publication information, users' experimental evaluation feedback and submitter's contact details. Gene expression assays are extended with a scalable assay viewer that provides detailed information on the alignment of primer and probe sequences on the known transcript variants of a gene, along with Single Nucleotide Polymorphisms (SNP) positions and peptide domain information. Furthermore, an mfold module is implemented to predict the secondary structure of the amplicon sequence, as this has been reported to impact the efficiency of the PCR. RTPrimerDB is also extended with an in silico analysis pipeline to streamline the evaluation of custom designed primer and probe sequences prior to ordering and experimental evaluation. In a secured environment, the pipeline performs automated BLAST specificity searches, mfold secondary structure prediction, SNP or plain sequence error identification, and graphical visualization of the aligned primer and probe sequences on the target gene.
- RTPrimerDB: the portal for real-time PCR primers and probes
Lefever S, Vandesompele J, Speleman F, Pattyn F.
Center for Medical Genetics, Ghent University Hospital, De Pintelaan 185, 9000 Gent, Belgium.
Nucleic Acids Res. 2009 Jan;37(Database issue): D942-945RTPrimerDB http://www.rtprimerdb.org is a freely accessible database and analysis tool for real-time quantitative PCR assays. RTPrimerDB includes records with user submitted assays that are linked to genome information from reference databases and quality controlled using an in silico assay evaluation system. The primer evaluation tools intended to assess the specificity and to detect features that could negatively affect the amplification efficiency are combined into a pipeline to test custom-designed primer and probe sequences. An improved user feedback system guides users and submitters to enter practical remarks and details about experimental evaluation analyses. The database is linked with reference databases to allow the submission of assays for all genes and organisms officially registered in Entrez Gene and RefSeq. Records in RTPrimerDB are assigned unique and stable identifiers. The content is provided via an interactive web-based search system and is available for download in the recently developed RDML format and as bulk export file. RTPrimerDB is a one-stop portal for high-quality and highly annotated real-time PCR assays.
- The BiSearch web server
Aranyi T, Varadi A, Simon I, Tusnady GE.
BMC Bioinformatics. 2006 ;7: 431.
lnstitute of Enzymology, BRC, HAS, H-1113 Karolina ut 29, Budapest, Hungary.BACKGROUND: A large number of PCR primer-design softwares are available online. However, only very few of them can be used for the design of primers to amplify bisulfite-treated DNA templates, necessary to determine genomic DNA methylation profiles. Indeed, the number of studies on bisulfite-treated templates exponentially increases as determining DNA methylation becomes more important in the diagnosis of cancers. Bisulfite-treated DNA is difficult to amplify since undesired PCR products are often amplified due to the increased sequence redundancy after the chemical conversion. In order to increase the efficiency of PCR primer-design, we have developed BiSearch web server, an online primer-design tool for both bisulfite-treated and native DNA templates. RESULTS: The web tool is composed of a primer-design and an electronic PCR (ePCR) algorithm. The completely reformulated ePCR module detects potential mispriming sites as well as undesired PCR products on both cDNA and native or bisulfite-treated genomic DNA libraries. Due to the new algorithm of the current version, the ePCR module became approximately hundred times faster than the previous one and gave the best performance when compared to other web based tools. This high-speed ePCR analysis made possible the development of the new option of high-throughput primer screening. BiSearch web server can be used for academic researchers at the http://bisearch.enzim.hu site. CONCLUSION: BiSearch web server is a useful tool for primer-design for any DNA template and especially for bisulfite-treated genomes. The ePCR tool for fast detection of mispriming sites and alternative PCR products in cDNA libraries and native or bisulfite-treated genomes are the unique features of the new version of BiSearch software.
-
BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes
Tusnady GE, Simon I, Varadi A, Aranyi T.
Nucleic Acids Res. 2005 Jan 13;33(1):e9.
Institute of Enzymology, BRC, Hungarian Academy of Sciences H-1113 Budapest, Karolina ut 29, Hungary.Bisulfite genomic sequencing is the most widely used technique to analyze the 5-methylation of cytosines, the prevalent covalent DNA modification in mammals. The process is based on the selective transformation of unmethylated cytosines to uridines. Then, the investigated genomic regions are PCR amplified, subcloned and sequenced. During sequencing, the initially unmethylated cytosines are detected as thymines. The efficacy of bisulfite PCR is generally low; mispriming and non-specific amplification often occurs due to the T richness of the target sequences. In order to ameliorate the efficiency of PCR, we developed a new primer-design software called BiSearch, available on the World Wide Web. It has the unique property of analyzing the primer pairs for mispriming sites on the bisulfite-treated genome and determines potential non-specific amplification products with a new search algorithm. The options of primer-design and analysis for mispriming sites can be used sequentially or separately, both on bisulfite-treated and untreated sequences. In silico and in vitro tests of the software suggest that new PCR strategies may increase the efficiency of the amplification.
- PRaTo: A web-tool to select optimal primer pairs for qPCR
Alberto Nonis, Marco Scortegagna, Alessandro Nonis, Benedetto Ruperti
Biochemical and Biophysical Research Communications 415 (2011) 707–708An essential pre-requisite to perform sound quantitative real-time polymerase chain reaction (qPCR) assays is to design outstanding primer pairs. This means they must have a good efficiency and be not prone to produce multiple amplicons or primer dimer products. To circumvent these issues, several softwares are available to help primer design. Although satisfactory computer-aided primer design tools are available for standard PCR, less efforts were done to provide specific methods for selection of optimal primer pairs for qPCR. We have developed PRaTo a web-based tool that enables checking and ranking of primers pairs for their attitude to perform optimally and reliably when used in qPCR experiments. PRaTo is available at http://prato.daapv.unipd.it
-
The ability to select short DNA oligonucleotide sequences capable of binding solely to their intended target is of great importance in developing nucleic acid based detection technologies. Applications such as multiplex PCR rely on primers binding to unique regions in a genome. Competing side reactions with other primer pairs or template DNA decrease PCR efficiency: Freely available primer design software such as Primer3 screens for potential hairpin and primer-dimer interactions while selecting a single primer pair. The development of multiplex PCR assays (in the range of 5 to 20 loci) requires the screening of all primer pairs for potential cross-reactivity. However, a logistical problem results due to the number of total number of comparisons required. Comparing the candidate oligomers rapidly for potential cross-reactivity reduces overall assay devlelopment time. Here we report the application of a familiar sliding algorithm for comparing two strands of DNA in an overlapping fashion. The algorithm has been employed in a software package wherein the user can compare user-defined threshold. Additional criteria of predicted melting temperature (Tm) and free energy of melting (deltaG) are included for further ranking. Sodium counterion and total stand concentrations can be adjusted for the Tm and deltaG calculations. primer set for a 10-plex assay (20 total primer sequences) results in 210 primer-primer combinations that must be screened. The ability to screen sets of multiple sequences in a single computational run. After the screening is completed, a score is assigned to potential duplex interactions exceeding a The predicted interactions are saved in a text file for further evaluation.
- A PCR primer bank for quantitative gene expression analysis
Wang X, Seed B.
Department of Molecular Biology, Massachusetts General Hospital, 50 Blossom Street, Boston, MA 02114, USA.
Nucleic Acids Res. 2003 Dec 15;31(24):e154Although gene expression profiling by microarray analysis is a useful tool for assessing global levels of transcriptional activity, variability associated with the data sets usually requires that observed differences be validated by some other method, such as real-time quantitative polymerase chain reaction (real-time PCR). However, non-specific amplification of non-target genes is frequently observed in the latter, confounding the analysis in approximately 40% of real-time PCR attempts when primer-specific labels are not used. Here we present an experimentally validated algorithm for the identification of transcript-specific PCR primers on a genomic scale that can be applied to real-time PCR with sequence-independent detection methods. An online database, PrimerBank, has been created for researchers to retrieve primer information for their genes of interest. PrimerBank currently contains 147 404 primers encompassing most known human and mouse genes. The primer design algorithm has been tested by conventional and real-time PCR for a subset of 112 primer pairs with a success rate of 98.2%.
- A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance
Spandidos A, Wang X, Wang H, Dragnev S, Thurber T, Seed B.
Center for Computational and Integrative Biology, Massachusetts General Hospital, MA, USA
BMC Genomics. 2008 Dec 24;9:633.BACKGROUND: Quantitative polymerase chain reaction (QPCR) is a widely applied analytical method for the accurate determination of transcript abundance. Primers for QPCR have been designed on a genomic scale but non-specific amplification of non-target genes has frequently been a problem. Although several online databases have been created for the storage and retrieval of experimentally validated primers, only a few thousand primer pairs are currently present in existing databases and the primers are not designed for use under a common PCR thermal profile.
RESULTS: We previously reported the implementation of an algorithm to predict PCR primers for most known human and mouse genes. We now report the use of that resource to identify 17483 pairs of primers that have been experimentally verified to amplify unique sequences corresponding to distinct murine transcripts. The primer pairs have been validated by gel electrophoresis, DNA sequence analysis and thermal denaturation profile. In addition to the validation studies, we have determined the uniformity of amplification using the primers and the technical reproducibility of the QPCR reaction using the popular and inexpensive SYBR Green I detection method.
CONCLUSION: We have identified an experimentally validated collection of murine primer pairs for PCR and QPCR which can be used under a common PCR thermal profile, allowing the evaluation of transcript abundance of a large number of genes in parallel. This feature is increasingly attractive for confirming and/or making more precise data trends observed from experiments performed with DNA microarrays.
- PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification
Spandidos A, Wang X, Wang H, Seed B.
Department of Genetics, Harvard Medical School, Center for Computational and Integrative Biology, Massachusetts General Hospital, Boston, MA 02114-2790, USA.
Nucleic Acids Res. 2010 Jan;38(Database issue): D792-799PrimerBank http://pga.mgh.harvard.edu/primerbank/ is a public resource for the retrieval of human and mouse primer pairs for gene expression analysis by PCR and Quantitative PCR (QPCR). A total of 306,800 primers covering most known human and mouse genes can be accessed from the PrimerBank database, together with information on these primers such as T(m), location on the transcript and amplicon size. For each gene, at least one primer pair has been designed and in many cases alternative primer pairs exist. Primers have been designed to work under the same PCR conditions, thus facilitating high-throughput QPCR. There are several ways to search for primers for the gene(s) of interest, such as by: GenBank accession number, NCBI protein accession number, NCBI gene ID, PrimerBank ID, NCBI gene symbol or gene description (keyword). In all, 26,855 primer pairs covering most known mouse genes have been experimentally validated by QPCR, agarose gel analysis, sequencing and BLAST, and all validation data can be freely accessed from the PrimerBank web site.
- PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update
Wang X, Spandidos A, Wang H, Seed B.
Department of Radiation Oncology, Washington University School of Medicine, 4511 Forest Park Ave, Saint Louis, MO 63108, USA.
Nucleic Acids Res. 2012 Jan;40(Database issue):D1144-9
Optimization of primer sequences for polymerase chain reaction (PCR) and quantitative PCR (qPCR) and reaction conditions remains an experimental challenge. We have developed a resource, PrimerBank, which contains primers that can be used for PCR and qPCR under stringent and allele-invariant amplification conditions. A distinguishing feature of PrimerBank is the experimental validation of primer pairs covering most known mouse genes. Here, we describe a major update of PrimerBank that includes the design of new primers covering 17,076 and 18,086 genes for the human and mouse species, respectively. As a result of this update, PrimerBank contains 497,156 primers (an increase of 62% from the previous version) that cover 36,928 human and mouse genes, corresponding to around 94% of all known protein-coding gene sequences. An updated algorithm based on our previous approach was used to design new primers using current genomic information available from the National Center for Biotechnology Information (NCBI). PrimerBank primers work under uniform PCR conditions, and can be used for high-throughput or genome-wide qPCR. Because of their broader linear dynamic range and greater sensitivity, qPCR approaches are used to reanalyze changes in expression suggested by exploratory technologies such as microarrays and RNA-Seq. The primers and all experimental validation data can be freely accessed from the PrimerBank website, http://pga.mgh.harvard.edu/primerbank/
Distribution of all rejected human primer candidates by various bioinformatics screening filters. Combined together, 99% of all primer candidates were rejected by these screening filters.

- GETPrime: a gene- or transcript-specific primer database for quantitative real-time PCR
Gubelmann C, Gattiker A, Massouras A, Hens K, David F, Decouttere F, Rougemont J, Deplancke B.
Institute of Bio-engineering, School of Life Sciences, Laboratory of Systems Biology and Genetics, Lausanne, Switzerland.
Database (Oxford). 2011 Sep 14;2011:bar040. Print 2011.
Database URL: http://deplanckelab.epfl.chThe vast majority of genes in humans and other organisms undergo alternative splicing, yet the biological function of splice variants is still very poorly understood in large part because of the lack of simple tools that can map the expression profiles and patterns of these variants with high sensitivity. High-throughput quantitative real-time polymerase chain reaction (qPCR) is an ideal technique to accurately quantify nucleic acid sequences including splice variants. However, currently available primer design programs do not distinguish between splice variants and also differ substantially in overall quality, functionality or throughput mode. Here, we present GETPrime, a primer database supported by a novel platform that uniquely combines and automates several features critical for optimal qPCR primer design. These include the consideration of all gene splice variants to enable either gene-specific (covering the majority of splice variants) or transcript-specific (covering one splice variant) expression profiling, primer specificity validation, automated best primer pair selection according to strict criteria and graphical visualization of the latter primer pairs within their genomic context. GETPrime primers have been extensively validated experimentally, demonstrating high transcript specificity in complex samples. Thus, the free-access, user-friendly GETPrime database allows fast primer retrieval and visualization for genes or groups of genes of most common model organisms, and is available at http://updepla1srv1.epfl.ch/getprime/
- methBLAST and methPrimerDB: web-tools for PCR based methylation analysis
Pattyn F, Hoebeeck J, Robbrecht P, Michels E, De Paepe A, Bottu G, Coornaert D, Herzog R, Speleman F, Vandesompele J.
Center for Medical Genetics, Ghent University Hospital, De Pintelaan 185, 9000 Ghent, Belgium.
BMC Bioinformatics. 2006 Nov 9;7:496.BACKGROUND: DNA methylation plays an important role in development and tumorigenesis by epigenetic modification and silencing of critical genes. The development of PCR-based methylation assays on bisulphite modified DNA heralded a breakthrough in speed and sensitivity for gene methylation analysis. Despite this technological advancement, these approaches require a cumbersome gene by gene primer design and experimental validation. Bisulphite DNA modification results in sequence alterations (all unmethylated cytosines are converted into uracils) and a general sequence complexity reduction as cytosines become underrepresented. Consequently, standard BLAST sequence homology searches cannot be applied to search for specific methylation primers.
RESULTS: To address this problem we developed methBLAST, a sequence similarity search program, based on the original BLAST algorithm but querying in silico bisulphite modified genome sequences to evaluate oligonucleotide sequence similarities. Apart from the primer specificity analysis tool, we have also developed a public database termed methPrimerDB for the storage and retrieval of validated PCR based methylation assays. The web interface allows free public access to perform methBLAST searches or database queries and to submit user based information. Database records can be searched by gene symbol, nucleotide sequence, analytical method used, Entrez Gene or methPrimerDB identifier, and submitter's name. Each record contains a link to Entrez Gene and PubMed to retrieve additional information on the gene, its genomic context and the article in which the methylation assay was described. To assure and maintain data integrity and accuracy, the database is linked to other reference databases. Currently, the database contains primer records for the most popular PCR-based methylation analysis methods to study human, mouse and rat epigenetic modifications. methPrimerDB and methBLAST are available at http://medgen.ugent.be/methprimerdb and http://medgen.ugent.be/methblast
CONCLUSION: We have developed two integrated and freely available web-tools for PCR based methylation analysis. methBLAST allows in silico assessment of primer specificity in PCR based methylation assays that can be stored in the methPrimerDB database, which provides a search portal for validated methylation assays.
- AtRTPrimer: database for Arabidopsis genome-wide homogeneous and specific RT-PCR primer-pairs
Han S, Kim D.
Department of BioSystems, Korea Advanced Institute of Science and Technology, 373-1 Guseong-dong, Yuseong-gu, Daejeon 305-701, Republic of Korea
BMC Bioinformatics. 2006 Mar 30;7:179.BACKGROUND: Primer design is a critical step in all types of RT-PCR methods to ensure specificity and efficiency of a target amplicon. However, most traditional primer design programs suggest primers on a single template of limited genetic complexity. To provide researchers with a sufficient number of pre-designed specific RT-PCR primer pairs for whole genes in Arabidopsis, we aimed to construct a genome-wide primer-pair database.
DESCRIPTION: We considered the homogeneous physical and chemical properties of each primer (homogeneity) of a gene, non-specific binding against all other known genes (specificity), and other possible amplicons from its corresponding genomic DNA or similar cDNAs (additional information). Then, we evaluated the reliability of our database with selected primer pairs from 15 genes using conventional and real time RT-PCR.
CONCLUSION: Approximately 97% of 28,952 genes investigated were finally registered in AtRTPrimer. Unlike other freely available primer databases for Arabidopsis thaliana, AtRTPrimer provides a large number of reliable primer pairs for each gene so that researchers can perform various types of RT-PCR experiments for their specific needs. Furthermore, by experimentally evaluating our database, we made sure that our database provides good starting primer pairs for Arabidopsis researchers to perform various types of RT-PCR experiments.
Real-time PCR primer design tools:
- RExPrimer: an integrated primer designing tool increases PCR effectiveness by avoiding 3' SNP-in-primer and mis-priming from structural variation
Piriyapongsa J, Ngamphiw C, Assawamakin A, Wangkumhang P, Suwannasri P, Ruangrit U, Agavatpanitch G, Tongsima S.
Genome Institute, National Center for Genetic Engineering and Biotechnology, Pathumthani, Thailand
BMC Genomics. 2009 Dec 3;10 Suppl 3:S4.
This software is freely available at http://www4a.biotec.or.th/rexprimerBACKGROUND: Polymerase chain reaction (PCR) is very useful in many areas of molecular biology research. It is commonly observed that PCR success is critically dependent on design of an effective primer pair. Current tools for primer design do not adequately address the problem of PCR failure due to mis-priming on target-related sequences and structural variations in the genome.
METHODS: We have developed an integrated graphical web-based application for primer design, called RExPrimer, which was written in Python language. The software uses Primer3 as the primer designing core algorithm. Locally stored sequence information and genomic variant information were hosted on MySQLv5.0 and were incorporated into RExPrimer.
RESULTS: RExPrimer provides many functionalities for improved PCR primer design. Several databases, namely annotated human SNP databases, insertion/deletion (indel) polymorphisms database, pseudogene database, and structural genomic variation databases were integrated into RExPrimer, enabling an effective without-leaving-the-website validation of the resulting primers. By incorporating these databases, the primers reported by RExPrimer avoid mis-priming to related sequences (e.g. pseudogene, segmental duplication) as well as possible PCR failure because of structural polymorphisms (SNP, indel, and copy number variation (CNV)). To prevent mismatching caused by unexpected SNPs in the designed primers, in particular the 3' end (SNP-in-Primer), several SNP databases covering the broad range of population-specific SNP information are utilized to report SNPs present in the primer sequences. Population-specific SNP information also helps customize primer design for a specific population. Furthermore, RExPrimer offers a graphical user-friendly interface through the use of scalable vector graphic image that intuitively presents resulting primers along with the corresponding gene structure. In this study, we demonstrated the program effectiveness in successfully generating primers for strong homologous sequences.
CONCLUSION: The improvements for primer design incorporated into RExPrimer were demonstrated to be effective in designing primers for challenging PCR experiments. Integration of SNP and structural variation databases allows for robust primer design for a variety of PCR applications, irrespective of the sequence complexity in the region of interest. This software is freely available at http://www4a.biotec.or.th/rexprimer
- BatchPrimer3: a high throughput web application for PCR and sequencing primer design
You FM, Huo N, Gu YQ, Luo MC, Ma Y, Hane D, Lazo GR, Dvorak J, Anderson OD.
Department of Plant Sciences, University of California, CA 95616, USA
BMC Bioinformatics. 2008 May 29;9:253.BACKGROUND: Microsatellite (simple sequence repeat - SSR) and single nucleotide polymorphism (SNP) markers are two types of important genetic markers useful in genetic mapping and genotyping. Often, large-scale genomic research projects require high-throughput computer-assisted primer design. Numerous such web-based or standard-alone programs for PCR primer design are available but vary in quality and functionality. In particular, most programs lack batch primer design capability. Such a high-throughput software tool for designing SSR flanking primers and SNP genotyping primers is increasingly demanded.
RESULTS: A new web primer design program, BatchPrimer3, is developed based on Primer3. BatchPrimer3 adopted the Primer3 core program as a major primer design engine to choose the best primer pairs. A new score-based primer picking module is incorporated into BatchPrimer3 and used to pick position-restricted primers. BatchPrimer3 v1.0 implements several types of primer designs including generic primers, SSR primers together with SSR detection, and SNP genotyping primers (including single-base extension primers, allele-specific primers, and tetra-primers for tetra-primer ARMS PCR), as well as DNA sequencing primers. DNA sequences in FASTA format can be batch read into the program. The basic information of input sequences, as a reference of parameter setting of primer design, can be obtained by pre-analysis of sequences. The input sequences can be pre-processed and masked to exclude and/or include specific regions, or set targets for different primer design purposes as in Primer3Web and primer3Plus. A tab-delimited or Excel-formatted primer output also greatly facilitates the subsequent primer-ordering process. Thousands of primers, including wheat conserved intron-flanking primers, wheat genome-specific SNP genotyping primers, and Brachypodium SSR flanking primers in several genome projects have been designed using the program and validated in several laboratories.
CONCLUSION: BatchPrimer3 is a comprehensive web primer design program to develop different types of primers in a high-throughput manner. Additional methods of primer design can be easily integrated into future versions of BatchPrimer3. The program with source code and thousands of PCR and sequencing primers designed for wheat and Brachypodium are accessible at http://wheat.pw.usda.gov/demos/BatchPrimer3/
- Java web tools for PCR, in silico PCR, and oligonucleotide assembly and analysis
Kalendar R, Lee D, Schulman AH.
MTT/BI Plant Genomics Laboratory, Institute of Biotechnology, University of Helsinki, P.O. Box 65, FIN-00014 Helsinki, Finland
Genomics. 2011 Aug;98(2):137-144The polymerase chain reaction is fundamental to molecular biology and is the most important practical molecular technique for the research laboratory. We have developed and tested efficient tools for PCR primer and probe design, which also predict oligonucleotide properties based on experimental studies of PCR efficiency. The tools provide comprehensive facilities for designing primers for most PCR applications and their combinations, including standard, multiplex, long-distance, inverse, real-time, unique, group-specific, bisulphite modification assays, Overlap-Extension PCR Multi-Fragment Assembly, as well as a programme to design oligonucleotide sets for long sequence assembly by ligase chain reaction. The in silico PCR primer or probe search includes comprehensive analyses of individual primers and primer pairs. It calculates the melting temperature for standard and degenerate oligonucleotides including LNA and other modifications, provides analyses for a set of primers with prediction of oligonucleotide properties, dimer and G-quadruplex detection, linguistic complexity, and provides a dilution and resuspension calculator.
- ConservedPrimers 2.0: a high-throughput pipeline for comparative genome referenced intron-flanking PCR primer design and its application in wheat SNP discovery
You FM, Huo N, Gu YQ, Lazo GR, Dvorak J, Anderson OD.
Department of Plant Sciences, University of California, Davis, CA 95616, USA
BMC Bioinformatics. 2009 Oct 13;10:331.BACKGROUND: In some genomic applications it is necessary to design large numbers of PCR primers in exons flanking one or several introns on the basis of orthologous gene sequences in related species. The primer pairs designed by this target gene approach are called "intron-flanking primers" or because they are located in exonic sequences which are usually conserved between related species, "conserved primers". They are useful for large-scale single nucleotide polymorphism (SNP) discovery and marker development, especially in species, such as wheat, for which a large number of ESTs are available but for which genome sequences and intron/exon boundaries are not available. To date, no suitable high-throughput tool is available for this purpose.
RESULTS: We have developed, the ConservedPrimers 2.0 pipeline, for designing intron-flanking primers for large-scale SNP discovery and marker development, and demonstrated its utility in wheat. This tool uses non-redundant wheat EST sequences, such as wheat contigs and singleton ESTs, and related genomic sequences, such as those of rice, as inputs. It aligns the ESTs to the genomic sequences to identify unique colinear exon blocks and predicts intron lengths. Intron-flanking primers are then designed based on the intron/exon information using the Primer3 core program or BatchPrimer3. Finally, a tab-delimited file containing intron-flanking primer pair sequences and their primer properties is generated for primer ordering and their PCR applications. Using this tool, 1,922 bin-mapped wheat ESTs (31.8% of the 6,045 in total) were found to have unique colinear exon blocks suitable for primer design and 1,821 primer pairs were designed from these single- or low-copy genes for PCR amplification and SNP discovery. With these primers and subsequently designed genome-specific primers, a total of 1,527 loci were found to contain one or more genome-specific SNPs.
CONCLUSION: The ConservedPrimers 2.0 pipeline for designing intron-flanking primers was developed and its utility demonstrated. The tool can be used for SNP discovery, genetic variation assays and marker development for any target genome that has abundant ESTs and a related reference genome that has been fully sequenced. The ConservedPrimers 2.0 pipeline has been implemented as a command-line tool as well as a web application. Both versions are freely available at http://wheat.pw.usda.gov/demos/ConservedPrimers/
- PriSM: a primer selection and matching tool for amplification and sequencing of viral genomes
Yu Q, Ryan EM, Allen TM, Birren BW, Henn MR, Lennon NJ.
Broad Institute of MIT & Harvard, Cambridge, MA 02142, USA.
Bioinformatics. 2011 Jan 15;27(2):266-7. Epub 2010 Nov 9.
AVAILABILITY: The program is freely available for use at: http://www.broadinstitute.org/perl/seq/specialprojects/primerDesign.cgiPriSM is a set of algorithms designed to select and match degenerate primer pairs for the amplification of viral genomes. The design of panels of hundreds of primer pairs takes just hours using this program, compared with days using a manual approach. PriSM allows for rapid in silico optimization of primers for downstream applications such as sequencing. As a validation, PriSM was used to create an amplification primer panel for human immunodeficiency virus (HIV) Clade B.
- PCRTiler: automated design of tiled and specific PCR primer pairs
Gervais AL, Marques M, Gaudreau L.
Département de Biologie, Université de Sherbrooke, 2500 boul. de l'Université, Sherbrooke, Qc, J1K 2R1, Canada
Nucleic Acids Res. 2010 Jul;38(Web Server issue): W308-312Efficiency and specificity of PCR amplification is dependent on several parameters, such as amplicon length, as well as hybridization specificity and melting temperature of primer oligonucleotides. Primer design is thus of critical importance for the success of PCR experiments, but can be a time-consuming and repetitive task, for example when large genomic regions are to be scanned for the presence of a protein of interest by chromatin immunoprecipitation experiments. We present here a webserver that allows the automated design of tiled primer pairs for any number of genomic loci. PCRTiler splits the target DNA sequences into smaller regions, and identifies candidate primers for each sub-region by running the well-known program Primer3 followed by the elimination of primers with a high cross-hybridization potential via BLAST. Tiling density and primer characteristics are specified by the user via a simple and user-friendly interface. The webserver can be accessed at http://pcrtiler.alaingervais.org:8080/PCRTiler Additionally, users may download a standalone Java-based implementation of this software. Experimental validation of PCRTiler has demonstrated that it produces correct results. We have tiled a region of the human genome, in which 96 of 123 primer pairs worked in the first attempt, and 105 of 123 (85%) could be made to work by optimizing the conditions of the PCR assay.
- maxAlike: maximum likelihood-based sequence reconstruction with application to improved primer design for unknown sequences
Menzel P, Stadler PF, Gorodkin J.
Center for non-coding RNA in Technology and Health, IBHV, University of Copenhagen, Grønnegårdsvej 3, DK-1870 Frederiksberg, Denmark.
Bioinformatics. 2011 Feb 1;27(3):317-25. Epub 2010 Dec 1.
AVAILABILITY: maxAlike is available for download and web server at: http://rth.dk/resources/maxAlikeMOTIVATION: The task of reconstructing a genomic sequence from a particular species is gaining more and more importance in the light of the rapid development of high-throughput sequencing technologies and their limitations. Applications include not only compensation for missing data in unsequenced genomic regions and the design of oligonucleotide primers for target genes in species with lacking sequence information but also the preparation of customized queries for homology searches.
RESULTS: We introduce the maxAlike algorithm, which reconstructs a genomic sequence for a specific taxon based on sequence homologs in other species. The input is a multiple sequence alignment and a phylogenetic tree that also contains the target species. For this target species, the algorithm computes nucleotide probabilities at each sequence position. Consensus sequences are then reconstructed based on a certain confidence level. For 37 out of 44 target species in a test dataset, we obtain a significant increase of the reconstruction accuracy compared to both the consensus sequence from the alignment and the sequence of the nearest phylogenetic neighbor. When considering only nucleotides above a confidence limit, maxAlike is significantly better (up to 10%) in all 44 species. The improved sequence reconstruction also leads to an increase of the quality of PCR primer design for yet unsequenced genes: the differences between the expected T(m) and real T(m) of the primer-template duplex can be reduced by ~26% compared with other reconstruction approaches. We also show that the prediction accuracy is robust to common distortions of the input trees. The prediction accuracy drops by only 1% on average across all species for 77% of trees derived from random genomic loci in a test dataset.
- PrimerIdent: A web based tool for conserved primer design
Pessoa AM, Pereira S, Teixeira J.
Bioinformation. 2010 Jul 6;5(2): 52-54
AVAILABILITY: http://primerident.up.ptConserved primers across multiple species and simultaneously specific for a certain isozyme can be rare and difficult to find. PrimerIdent was developed aiming to automate this primer design and selection process in a given nucleotide sequence alignment, providing an intuitive, easy to interpret graphical result, which offers a list of all possible primers that meet the user criteria, with a colour-code identity to each sequence in the alignment. The software here presented is a simple and intuitive web based tool that is suitable for distinguishing very similar nucleotide sequences, such as isozymes-coding sequences, to enable the conserved primer design across multiple species, necessary for approaches that rely on knowing if a primer is suitable for a certain set of pre-aligned sequences, to design a specific primer to a certain sequence variation, or a combination thereof. This extremely useful software can, therefore, be used as a tool for the specific amplification of individual members of multigenic families across related species and also to evaluate the differential expression of isogenes for a given species.
- ThermoPhyl: a software tool for selecting phylogenetically optimized conventional and quantitative-PCR taxon-targeted assays for use with complex samples
Oakley BB, Dowd SE, Purdy KJ.
University of Warwick, School of Life Sciences, Coventry, UK
FEMS Microbiol Ecol. 2011 Jul;77(1): 17-27The ability to specifically and sensitively target genotypes of interest is critical for the success of many PCR-based analyses of environmental or clinical samples that contain multiple templates. Next-generation sequence data clearly show that such samples can harbour hundreds to thousands of operational taxonomic units, a richness that precludes the manual evaluation of candidate assay specificity and sensitivity using multiple sequence alignments. To solve this problem, we have developed and validated a free software tool that automates the identification of PCR assays targeting specific genotypes in complex samples. ThermoPhyl uses user-defined target and nontarget sequence databases to assess the phylogenetic sensitivity and specificity of thermodynamically optimized candidate assays derived from primer design software packages. ThermoPhyl derives its name from its central premise of testing Thermodynamically optimal assays for Phylogenetic specificity and sensitivity and can be used for two primer (traditional PCR) or two primers with an internal probe (e.g. TaqMan(®) qPCR) application and potentially for oligonucleotide probes. Here, we describe the use of ThermoPhyl for traditional PCR and qPCR assays. PCR assays selected using ThermoPhyl were validated using 454 pyrosequencing of a traditional specific PCR assay and with a set of four genotype-specific qPCR assays applied to estuarine sediment samples.
- WASP: a Web-based Allele-Specific PCR assay designing tool for detecting SNPs and mutations
Wangkumhang P, Chaichoompu K, Ngamphiw C, Ruangrit U, Chanprasert J, Assawamakin A, Tongsima S.
Biostatistics and Informatics Laboratory, Genomics Institute, National Center for Genetic Engineering and Biotechnology, Thailand Science Park, Pathumtani, Thailand
BMC Genomics. 2007 Aug 14;8:275.BACKGROUND: Allele-specific (AS) Polymerase Chain Reaction is a convenient and inexpensive method for genotyping Single Nucleotide Polymorphisms (SNPs) and mutations. It is applied in many recent studies including population genetics, molecular genetics and pharmacogenomics. Using known AS primer design tools to create primers leads to cumbersome process to inexperience users since information about SNP/mutation must be acquired from public databases prior to the design. Furthermore, most of these tools do not offer the mismatch enhancement to designed primers. The available web applications do not provide user-friendly graphical input interface and intuitive visualization of their primer results.
RESULTS: This work presents a web-based AS primer design application called WASP. This tool can efficiently design AS primers for human SNPs as well as mutations. To assist scientists with collecting necessary information about target polymorphisms, this tool provides a local SNP database containing over 10 million SNPs of various populations from public domain databases, namely NCBI dbSNP, HapMap and JSNP respectively. This database is tightly integrated with the tool so that users can perform the design for existing SNPs without going off the site. To guarantee specificity of AS primers, the proposed system incorporates a primer specificity enhancement technique widely used in experiment protocol. In particular, WASP makes use of different destabilizing effects by introducing one deliberate 'mismatch' at the penultimate (second to last of the 3'-end) base of AS primers to improve the resulting AS primers. Furthermore, WASP offers graphical user interface through scalable vector graphic (SVG) draw that allow users to select SNPs and graphically visualize designed primers and their conditions.
CONCLUSION: WASP offers a tool for designing AS primers for both SNPs and mutations. By integrating the database for known SNPs (using gene ID or rs number), this tool facilitates the awkward process of getting flanking sequences and other related information from public SNP databases. It takes into account the underlying destabilizing effect to ensure the effectiveness of designed primers. With user-friendly SVG interface, WASP intuitively presents resulting designed primers, which assist users to export or to make further adjustment to the design. This software can be freely accessed at http://bioinfo.biotec.or.th/WASP
About QuantPrime - http://www.quantprime.de
QuantPrime is an intuitive and user-friendly, fully automated tool for primer pair design in small- to large-scale real-time reverse transcription qPCR (also known as realtime qRT-PCR or RT-qPCR) analyses. QuantPrime can be used on the website or on a local computer (contact us for getting a copy); it offers design and specificity checking with highly customizable parameters and is ready to use with most publicly available eukaryotic transcriptomes, including all higher eukaryote model organisms and important plant crops, while benefiting from exon-intron border and splice variant information in available genome annotations. Experimental results with the model plant Arabidopsis thaliana, the crop Hordeum vulgare (barley) and the model green alga Chlamydomonas reinhardtii show success rates of designed primer pairs exceeding 96 %. For more information on the algorithms used in QuantPrime, please read the paper published in BMC Bioinformatics: QuantPrime - a flexible tool for reliable high-throughput primer design for quantitative PCR.
The QuantPrime service was created and is being maintained by Samuel Arvidsson (supported by EU contract MRTN-CT-2006-035833) at the University of Potsdam. The graphical design on the web site was created by Dr. Mirosław Kwaśniewski, who also helped out with the design of the program. The server is administrated by Diego Mauricio Riaño-Pachón, who helped out with the design of the program. The work is supervised by Prof. Dr. Bernd Müller-Röber.
We kindly ask QuantPrime users for citation when primers are used in published works. Please cite as follows:
QuantPrime - a flexible tool for reliable high-throughput primer design for quantitative PCR.
Arvidsson S, Kwasniewski M, Riaño-Pachón DM, Mueller-Roeber B.
Max-Planck Institute of Molecular Plant Physiology, Potsdam-Golm, Germany
BMC Bioinformatics. 2008 9: 465
BACKGROUND: Medium- to large-scale expression profiling using quantitative polymerase chain reaction (qPCR) assays are becoming increasingly important in genomics research. A major bottleneck in experiment preparation is the design of specific primer pairs, where researchers have to make several informed choices, often outside their area of expertise. Using currently available primer design tools, several interactive decisions have to be made, resulting in lengthy design processes with varying qualities of the assays.
RESULTS: Here we present QuantPrime, an intuitive and user-friendly, fully automated tool for primer pair design in small- to large-scale qPCR analyses. QuantPrime can be used online through the internet http://www.quantprime.de/ or on a local computer after download; it offers design and specificity checking with highly customizable parameters and is ready to use with many publicly available transcriptomes of important higher eukaryotic model organisms and plant crops (currently 295 species in total), while benefiting from exon-intron border and alternative splice variant information in available genome annotations. Experimental results with the model plant Arabidopsis thaliana, the crop Hordeum vulgare and the model green alga Chlamydomonas reinhardtii show success rates of designed primer pairs exceeding 96%.
CONCLUSION: QuantPrime constitutes a flexible, fully automated web application for reliable primer design for use in larger qPCR experiments, as proven by experimental data. The flexible framework is also open for simple use in other quantification applications, such as hydrolyzation probe design for qPCR and oligonucleotide probe design for quantitative in situ hybridization. Future suggestions made by users can be easily implemented, thus allowing QuantPrime to be developed into a broad-range platform for the design of RNA expression assays.
Human Endogenous Control Gene Panel
For all gene expression studies using quantitative PCR it is necessary to compensate for differences between samples due to material losses, differences in RT yields and PCR inhibition. Normalization should include an endogenous control gene, but can also be complemented by identical sample input amounts. The endogenous control gene should have constant expression in all the samples compared. There is no universal control gene, expressed at a constant level under all conditions and in all tissues.
The best way to choose the proper reference gene is by running a panel of potential genes on a number of representative test samples. The gene(s) most appropriate for normalization are chosen in each case.
The Human Endogenous Control Panel consists of 12 validated qPCR assays for the most common endogenous control genes for gene expression studies, and provides a rapid and cost efficient way to identify your control genes. The panel is compatible with most commercial mastermixes containg SYBR Green I => short manual
PCR Primer Design Guidelines 
Polymerase Chain Reaction is widely held as one of the most important inventions of the 20th century in molecular biology. Small amounts of the genetic material can now be amplified to be able to a identify, manipulate DNA, detect infectious organisms, including the viruses that cause AIDS, hepatitis, tuberculosis, detect genetic variations, including mutations, in human genes and numerous other tasks.
PCR involves the following three steps: denaturation, annealing and extension. First, the genetic material is denatured, converting the double stranded DNA molecules to single strands. The primers are then annealed to the complementary regions of the single stranded molecules. In the third step, they are extended by the action of the DNA polymerase. All these steps are temperature sensitive and the common choice of temperatures is 94oC, 60oC and 70oC respectively. Good primer design is essential for successful reactions. The important design considerations described below are a key to specific amplification with high yield. The preferred values indicated are built into all our products by default.
1. Primer Length: It is generally accepted that the optimal length of PCR primers is 18-22 bp. This length is long enough for adequate specificity, and short enough for primers to bind easily to the template at the annealing temperature.
2. Primer Melting Temperature: Primer Melting Temperature (Tm) by definition is the temperature at which one half of the DNA duplex will dissociate to become single stranded and indicates the duplex stability. Primers with melting temperatures in the range of 52-58 oC generally produce the best results. Primers with melting temperatures above 65oC have a tendency for secondary annealing. The GC content of the sequence gives a fair indication of the primer Tm. All our products calculate it using the nearest neighbor thermodynamic theory, accepted as a much superior method for estimating it, which is considered the most recent and best available.
Formula for primer Tm calculation:
Melting Temperature Tm(oK) = {ΔH/ ΔS + R ln(C)}, Or Melting Temperature Tm(oC) = {ΔH/ ΔS + R ln(C)} - 273.15 where
ΔH (kcal/mole) : H is the Enthalpy. Enthalpy is the amount of heat energy possessed by substances. ΔH is the change in Enthalpy. In the above formula the ΔH is obtained by adding up all the di-nucleotide pairs enthalpy values of each nearest neighbor base pair.
ΔS (kcal/mole) : S is the amount of disorder a system exhibits is called entropy. ΔS is change in Entropy. Here it is obtained by adding up all the di-nucleotide pairs entropy values of each nearest neighbor base pair. An additional salt correction is added as the Nearest Neighbor parameters were obtained from DNA melting studies conducted in 1M Na+ buffer and this is the default condition used for all calculations.
ΔS (salt correction) = ΔS (1M NaCl )+ 0.368 x N x ln([Na+])
Where
N is the number of nucleotide pairs in the primer ( primer length -1).
[Na+] is salt equivalent in mM.
[Na+] calculation:
[Na+] = Monovalent ion concentration +4 x free Mg2+
3.Primer annealing temperature : The primer melting temperature is the estimate of the DNA-DNA hybrid stability and critical in determining the annealing temperature. Too high Ta will produce insufficient primer-template hybridization resulting in low PCR product yield. Too low Ta may possibly lead to non-specific products caused by a high number of base pair mismatches,. Mismatch tolerance is found to have the strongest influence on PCR specificity.
Ta = 0.3 x Tm(primer) + 0.7 Tm (product) – 14.9
where, T m (primer) = Melting Temperature of the primers
Tm(product) = Melting temperature of the product
4. GC Content : The GC content (the number of G's and C's in the primer as a percentage of the total bases) of primer should be 40-60%.
5. GC Clamp : The presence of G or C bases within the lat five bases from the 3' end of primers (GC clamp) helps promote specific binding at the 3' end due to the stronger bonding of G and C bases. More than 3 G's or C's should be avoided in the last 5 bases at the 3' end of the primer.
6. Primer Secondary Structures : Presence of the primer secondary structures produced by intermolecular or intramolecular interactions can lead to poor or no yield of the product. They adversely affect primer template annealing and thus the amplification. They greatly reduce the availability of primers to the reaction.
i) Hairpins : It is formed by intramolecular interaction within the primer and should be avoided. Optimally a 3' end hairpin with a ΔG of -2 kcal/mol and an internal hairpin with a ΔG of -3 kcal/mol is tolerated generally.

ΔG definition : The Gibbs Free Energy G is the measure of the amount of work that can be extracted from a process operating at a constant pressure. It is the measure of the spontaneity of the reaction. The stability of hairpin is commonly represented by its ΔG value, the energy required to break the secondary structure. Larger negative value for ΔG indicates stable, undesirable hairpins. Presence of hairpins at the 3' end most adversely affects the reaction.
ΔG = ΔH – TΔS
ii) Self Dimer : A primer self-dimer is formed by intermolecular interactions between the two (same sense) primers, where the primer is homologous to itself. Generally a large amount of primers are used in PCR compared to the amount of target gene. When primers form intermolecular dimers much more readily than hybridizing to target DNA, they reduce the product yield. Optimally a 3' end self dimer with a ΔG of -5 kcal/mol and an internal self dimer with a ΔG of -6 kcal/mol is tolerated generally.
iii) Cross Dimer : Primer cross dimers are formed by intermolecular interaction between sense and antisense primers, where they are homologous. Optimally a 3' end cross dimer with a ΔG of -5 kcal/mol and an internal cross dimer with a ΔG of -6 kcal/mol is tolerated generally.

7. Repeats : A repeat is a di-nucleotide occurring many times consecutively and should be avoided because they can misprime. For example: ATATATAT. A maximum number of di-nucleotide repeats acceptable in an oligo is 4 di-nucleotides.
8. Runs : Primers with long runs of a single base should generally be avoided as they can misprime. For example, AGCGGGGGATGGGG has runs of base 'G' of value 5 and 4. A maximum number of runs accepted is 4bp.
9. 3' End Stability : It is the maximum ΔG value of the five bases from the 3' end. An unstable 3' end (less negative ΔG) will result in less false priming.
10. Avoid Template secondary structure : A single stranded Nucleic acid sequences is highly unstable and fold into conformations (secondary structures). The stability of these template secondary structures depends largely on their free energy and melting temperatures(Tm). Consideration of template secondary structures is important in designing primers, especially in qPCR. If primers are designed on a secondary structures which is stable even above the annealing temperatures, the primers are unable to bind to the template and the yield of PCR product is significantly affected. Hence, it is important to design primers in the regions of the templates that do not form stable secondary structures during the PCR reaction. Our products determine the secondary structures of the template using the Mfold algorithm and design primers avoiding them.
11. Avoid Cross homology : To improve specificity of the primers it is necessary to avoid regions of homology. Primers designed for a sequence must not amplify other genes in the mixture. Commonly, primers are designed and then BLASTed to test the specificity. Our products offer a better alternative. You can avoid regions of cross homology while designing primers. You can BLAST the templates against the appropriate non-redundant database and the software will interpret the results. It will identify regions significant cross homologies in each template and avoid them during primer search.
Parameters for Primer Pair Design:
1. Amplicon Length : The amplicon length is dictated by the experimental goals. For qPCR, the target length is closer to 100 bp and for standard PCR, it is near 500 bp. If you know the positions of each primer with respect to the template, the product is calculated as: Product length = (Position of antisense primer-Position of sense primer) + 1.
2. Product position : Primer can be located near the 5' end, the 3' end or any where within specified length. Generally, the sequence close to the 3' end is known with greater confidence and hence preferred most frequently.
3. Tm of Product : Melting Temperature (Tm) is the temperature at which one half of the DNA duplex will dissociate and become single stranded. The stability of the primer-template DNA duplex can be measured by the melting temperature (Tm).
4.Optimum Annealing temperature (Ta Opt): The formula of Rychlik is most respected. Our products use this formula to calculate it and thousands of our customers have reported good results using it for the annealing step of the PCR cycle. It usually results in good PCR product yield with minimum false product production.
T a Opt = 0.3 x(T m of primer) + 0.7 x(T m of product) - 25
where
Tm of primer is the melting temperature of the less stable primer-template pair
Tm of product is the melting temperature of the PCR product.
5. Primer Pair Tm Mismatch Calculation : The two primers of a primer pair should have closely matched melting temperatures for maximizing PCR product yield. The difference of 5oC or more can lead no amplification.
Primer Design Using Software
A number of primer design tools are available that can assist in PCR primer design for new and experienced users alike. These tools may reduce the cost and time involved in experimentation by lowering the chances of failed experimentation.
Primer Premier follows all the guidelines specified for PCR primer design. Primer Premier can be used to design primers for single templates, alignments, degenerate primer design, restriction enzyme analysis. contig analysis and design of sequencing primers.
The guidelines for qPCR primer design vary slightly. Software such as AlleleID and Beacon Designer can design primers and oligonucleotide probes for complex detection assays such as multiplex assays, cross species primer design, species specific primer design and primer design to reduce the cost of experimentation.
PrimerPlex is a software that can design ASPE (Allele specific Primer Extension) primers and capture probes for multiplex SNP genotyping using suspension array systems such as Luminex xMAP® and BioRad Bioplex.
References :
1. "A critical review of PCR primer design algorithms and cross-hybridization case study" By F.John Burpo.
2. "Optimization of the annealing temperature for DNA amplification in vitro" By W.Rychlik, W.J.Spencer
and R.E.Rhoads.
3. "A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics" By John SantaLucia.
4. "A computer program for selection of oligonucleotide primers for polymerase chain reactions" Lowe T, Sharefkin J, Yang SQ, Dieffenbach CW.
5. "Optimization strategies for the polymerase chain reaction" Williams JF.Perkin-Elmer Corporation, Norwalk, CT 06859-0251.
6. "Algorithms and thermodynamics for RNA secondary structure prediction. A Practical guide." Zuker.m.athews, D.Turner, D.
Summary of Primer Design Softwares:
| |||||
|
For routine needs, improve your assay with our OligoArchitect online design tool powered by the industry standard Beacon Designer™ (Premier Biosoft). The user-friendly interface utilizes the latest algorithms, provides results in real time, supports templates up to 10,000 base pairs, and allows for the adjustment of input parameters such as homopolymer run/repeat maximum length, G/C clamp length, and maximum primer pair Tm mismatch. Designs can be completed for traditional PCR or quantitative real-time PCR (qPCR) using the following detection chemistries:
Our online design tool can be used for the following applications:
OligoArchitect online is easy to use: select either 'SYBR Green' or 'Dual Labeled Probe', type in the name of your assay, paste in your sequence, and click search. You can add and delete a SNP or select 'Primer Parameter' if you wish to adjust the default settings. Download the glossary (387 Kb PDF) to learn more about the various parameters. All reported sequences, associated properties, and assay parameters are available for export to Excel and convenient email ordering. To establish MIQE-compliance, be sure to
| |||||
| Assay Design Tools for qPCR IDT offers a group of qPCR design tools to suit your specific application needs. Whether you want to quickly order a predesigned assay, or desire assistance with designing assays for a challenging target, IDT has a design tool that can help. With three different programs to choose from, it is important to know which will provide the optimal qPCR assay for your needs. Each of the three design programs requires only the NCBI RefSeq number, which can be used to pull the target sequence from NCBI. If you are working with unannotated sequences, the freely available RealTime and PrimerQuestSM programs can also design assays for a user-specified target. Key Features of Assay Design Tools PrimeTime® qPCR Program
PrimerQuestSM PrimerQuest is a useful tool for qPCR assay designs with non-standard requirements. This program is not specific for qPCR design like our other two programs, but if your design requires more demanding customization, this highly flexible program can be of great use. PrimerQuest designs can be customized in many ways, such as directing the assay towards certain areas of your target or by specifying primer or probe sequences. IDT Technical Support can also offer assistance with this program to help you meet your specific design challenges. | |||||
| PrimeSyn Lab | |||||
| PCR Primer Design Guidelines | |||||
| PRaTo, a web-tool for the selection of the best primer pair for qPCR http://prato.daapv.unipd.it PRaTo: A web-tool to select optimal primer pairs for qPCR An essential pre-requisite to perform sound quantitative real-time polymerase chain reaction (qPCR) assays is to design outstanding primer pairs. This means they must have a good efficiency and be not prone to produce multiple amplicons or primer dimer products. To circumvent these issues, several softwares are available to help primer design. Although satisfactory computer-aided primer design tools are available for standard PCR, less efforts were done to provide specific methods for selection of optimal primer pairs for qPCR. We have developed PRaTo a web-based tool that enables checking and ranking of primers pairs for their attitude to perform optimally and reliably when used in qPCR experiments. PRaTo is available at http://prato.daapv.unipd.it | |||||
| Design of Primers for Automated Sequencing One of the most important factors in successful automated DNA sequencing is proper primer design. This document describes the steps involved in this process and the major pitfalls to avoid. **** Use a Computer to Design Primers **** We highly recommend that a computer be used during primer design in order to check for certain fatal design flaws. Numerous programs are capable of performing this analysis. For example, look for 'Primer3' on the web. Some Basic Concepts: If you are confused by the strands and primer orientation, read this. Sequencing primers must be able to anneal to the target DNA in a predictable location and on a predictable strand. They furthermore must be capable of extension by Taq DNA Polymerase. Some people are confused about how to examine a DNA sequence to choose an appropriate primer sequence. Here are a few things for novices to remember:
| |||||
| Alkami Biosystems Quick Guide for PCR This 158 page PCR manual covers the following topics: PCR Primer design, PCR Methods, PCR Polymerases, PCR Variables, PCR Troubleshooting, Special PCR Topics, Appendixes and a comprehensive Index. This FREE online guide is in PDF format. | |||||
| ExonPrimer is a Perl script that helps to design intronic primers for the PCR amplification of exons. The script needs a cDNA and the corresponding genomic sequence as input. It aligns these sequences using Blat and designs PCR primers to amplify each exon using Primer3. The positions of the exons are deduced from the alignment of the genomic and the cDNA sequences. Insertions/deletions up to 6 base pairs are bridged by postprocessing. Exons with small introns in-between are combined. Exons smaller than 20-25 bp will not be recognized. The user can define the maximum exon size. Exons larger than this size will be divided into several parts. The poly-A tail of the cDNA should be clipped to allow the alignment of the cDNA and the genomic DNA sequence. The genomic sequence must be longer than the cDNA sequence. Otherwise the design of primers for the first and/or last exon is not possible. Download of the human genome sequence with all SNPs masked by N's. Using this sequence, one can avoid primers to be positioned across SNPs. ExonPrimer is also available in the UCSC Genome Browser for the human genome assemblies hg16 (July 2003) and hg17 (May 2004). One can find a link to ExonPrimer in the 'Quick Links to Tools and Databases' section of the known genes details page. | |||||
| Design of Primers for Automated Sequencing | |||||
| GeneFisher (manual pages) Interactive Primer Design ( Folker Meyer & Chris Schleiermacher) | |||||




Allele Specific Pcr Primer Design Tool
Source: https://www.gene-quantification.de/primers.html
Posted by: marshallknowded.blogspot.com
0 Response to "Allele Specific Pcr Primer Design Tool"
Post a Comment